Navigating the pitfalls of applying machine learning in genomics
The scale of genetic, epigenomic, transcriptomic, cheminformatic and proteomic data available today, coupled with easy-to-use machine learning (ML) toolkits, has propelled the application of supervised learning in genomics research. However, the assumptions behind the statistical models and performance evaluations in ML software frequently are not met in biological systems. In this Review, we illustrate the impact of several common pitfalls encountered when applying supervised ML in genomics. We explore how the structure of genomics data can bias performance evaluations and predictions. To address the challenges associated with applying cutting-edge ML methods to genomics, we describe solutions and appropriate use cases where ML modelling shows great potential.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
206,07 € per year
only 17,17 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others
A self-supervised deep learning method for data-efficient training in genomics
Article Open access 11 September 2023
Applying interpretable machine learning in computational biology—pitfalls, recommendations and opportunities for new developments
Article 09 August 2024

Essentiality, protein–protein interactions and evolutionary properties are key predictors for identifying cancer-associated genes using machine learning
Article Open access 22 April 2024
References
- Teschendorff, A. E. Avoiding common pitfalls in machine learning omic data science. Nat. Mater.18, 422–427 (2019). This Comment article talks about cross-validation and independent test sets as solutions to two pitfalls encountered when applying supervised ML in genomics: the ‘curse of dimensionality’ and confounding. ArticleCASPubMedGoogle Scholar
- Minhas, F., Asif, A. & Ben-Hur, A. Ten ways to fool the masses with machine learning. Preprint at arXivhttps://arxiv.org/abs/1901.01686 (2019).
- Eraslan, G., Avsec, Ž., Gagneur, J. & Theis, F. J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet.20, 389–403 (2019). ArticleCASPubMedGoogle Scholar
- Ching, T. et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface15, 20170387 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Zou, J. et al. A primer on deep learning in genomics. Nat. Genet.51, 12–18 (2019). ArticleCASPubMedGoogle Scholar
- Flagel, L., Brandvain, Y. & Schrider, D. R. The unreasonable effectiveness of convolutional neural networks in population genetic inference. Mol. Biol. Evol.36, 220–238 (2019). ArticleCASPubMedGoogle Scholar
- Liu, J., Lewinger, J. P., Gilliland, F. D., Gauderman, W. J. & Conti, D. V. Confounding and heterogeneity in genetic association studies with admixed populations. Am. J. Epidemiol.177, 351–360 (2013). ArticlePubMedPubMed CentralGoogle Scholar
- Vilhjálmsson, B. J. & Nordborg, M. The nature of confounding in genome-wide association studies. Nat. Rev. Genet.14, 1–2 (2013). ArticlePubMedGoogle Scholar
- Hellwege, J. N. et al. Population stratification in genetic association studies. Curr. Protoc. Hum. Genet.95, 1.22.1–1.22.23 (2017). Google Scholar
- Sul, J. H., Martin, L. S. & Eskin, E. Population structure in genetic studies: confounding factors and mixed models. PLoS Genet.14, e1007309 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Weirauch, M. T. et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol.31, 126–134 (2013). ArticleCASPubMedPubMed CentralGoogle Scholar
- Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet.11, 733–739 (2010). This Review documents the prevalence of batch effects in genomic data and shows how these can confound statistical inferences. ArticleCASPubMedGoogle Scholar
- Tran, H. T. N. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol.21, 12 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Rabanser, S., Günnemann, S. & Lipton, Z. Failing loudly: an empirical study of methods for detecting dataset shift. in Advances in Neural Information Processing Systems (NeurIPS 2019) (eds Wallach, H. et al.) Vol. 32, 1396–1408 (Curran Associates, Inc., 2019).
- Gretton, A., Borgwardt, K. M., Rasch, M. J., Schölkopf, B. & Smola, A. A kernel two-sample test. J. Mach. Learn. Res.13, 723–773 (2012). Google Scholar
- Ren, J. et al. in Advances in Neural Information Processing Systems (NeurIPS 2019) (eds Wallach, H. et al.) Vol. 32, 14707–14718 (Curran Associates, Inc., 2019).
- Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. Preprint at arXivhttps://arxiv.org/abs/1312.6114# (2013).
- Liu, F. T., Ting, K. M. & Zhou, Z. in IEEE International Conference on Data Mining 413–422 (IEEE, 2008).
- Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics8, 118–127 (2007). ArticlePubMedGoogle Scholar
- Leek, J. T. & Storey, J. D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet.3, 1724–1735 (2007). ArticleCASPubMedGoogle Scholar
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol.36, 411–420 (2018). ArticleCASPubMedPubMed CentralGoogle Scholar
- Stuart, T. et al. Comprehensive integration of single-cell data. Cell177, 1888–1902.e21 (2019). ArticleCASPubMedPubMed CentralGoogle Scholar
- Wang, T. et al. BERMUDA: a novel deep transfer learning method for single-cell RNA sequencing batch correction reveals hidden high-resolution cellular subtypes. Genome Biol.20, 165 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Pan, S. J. & Yang, Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng.22, 1345–1359 (2010). ArticleGoogle Scholar
- Kouw, W. M. & Loog, M. A review of domain adaptation without target labels. IEEE Trans. Pattern Anal. Mach. Intell.43, 766–785 (2019). ArticleGoogle Scholar
- Shimodaira, H. Improving predictive inference under covariate shift by weighting the log-likelihood function. J. Stat. Plan. Inference90, 227–244 (2000). This paper discusses distributional differences, also known as covariate shift, and proposes several weighting schemes for adjusting for this pitfall. ArticleGoogle Scholar
- Bickel, S., Brückner, M. & Scheffer, T. Discriminative learning under covariate shift. J. Mach. Learn. Res.10, 2137–2155 (2009). Google Scholar
- Orenstein, Y. & Shamir, R. Modeling protein-DNA binding via high-throughput in vitro technologies. Brief. Funct. Genomics16, 171–180 (2017). CASPubMedGoogle Scholar
- Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol.33, 831–838 (2015). ArticleCASPubMedGoogle Scholar
- Berger, M. F. & Bulyk, M. L. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc.4, 393–411 (2009). ArticleCASPubMedPubMed CentralGoogle Scholar
- Annala, M., Laurila, K., Lähdesmäki, H. & Nykter, M. A linear model for transcription factor binding affinity prediction in protein binding microarrays. PLoS ONE6, e20059 (2011). ArticleCASPubMedPubMed CentralGoogle Scholar
- Agius, P., Arvey, A., Chang, W., Noble, W. S. & Leslie, C. High resolution models of transcription factor-DNA affinities improve in vitro and in vivo binding predictions. PLoS Comput. Biol.6, e1000916 (2010). ArticlePubMedPubMed CentralGoogle Scholar
- Riley, T. R., Lazarovici, A., Mann, R. S. & Bussemaker, H. J. Building accurate sequence-to-affinity models from high-throughput in vitro protein-DNA binding data using FeatureREDUCE. Elife4, e06397 (2015). ArticlePubMedPubMed CentralGoogle Scholar
- Wong, K.-C., Li, Y., Peng, C. & Wong, H.-S. A comparison study for DNA motif modeling on protein binding microarray. IEEE/ACM Trans. Comput. Biol. Bioinform.13, 261–271 (2016). ArticleCASPubMedGoogle Scholar
- Rastogi, C. et al. Accurate and sensitive quantification of protein-DNA binding affinity. Proc. Natl Acad. Sci. USA115, E3692–E3701 (2018). ArticleCASPubMedPubMed CentralGoogle Scholar
- Im, J., Park, B. & Han, K. A generative model for constructing nucleic acid sequences binding to a protein. BMC Genomics20, 967 (2019). ArticleCASPubMedPubMed CentralGoogle Scholar
- Ishida, R. et al. RaptRanker: in silico RNA aptamer selection from HT-SELEX experiment based on local sequence and structure information. Nucleic Acids Res.48, e82 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Nutiu, R. et al. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nat. Biotechnol.29, 659–664 (2011). ArticleCASPubMedPubMed CentralGoogle Scholar
- Tabb, D. L. et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res.9, 761–776 (2010). ArticleCASPubMedPubMed CentralGoogle Scholar
- Pooch, E. H. P., Ballester, P. L. & Barros, R. C. Can we trust deep learning models diagnosis? The impact of domain shift in chest radiograph classification. Preprint at arXivhttps://arxiv.org/abs/1909.01940# (2019).
- Zech, J. R. et al. Variable generalization performance of a deep learning model to detect pneumonia in chest radiographs: a cross-sectional study. PLoS Med.15, e1002683 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Badgeley, M. A. et al. Deep learning predicts hip fracture using confounding patient and healthcare variables. NPJ Digit. Med.2, 31 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Antun, V., Renna, F., Poon, C., Adcock, B. & Hansen, A. C. On instabilities of deep learning in image reconstruction and the potential costs of AI. Proc. Natl Acad. Sci. USA117, 30088–30095 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Geis, J. R. et al. Ethics of artificial intelligence in radiology: summary of the joint european and north american multisociety statement. Radiology293, 436–440 (2019). ArticlePubMedGoogle Scholar
- Larrazabal, A. J., Nieto, N., Peterson, V., Milone, D. H. & Ferrante, E. Gender imbalance in medical imaging datasets produces biased classifiers for computer-aided diagnosis. Proc. Natl Acad. Sci. USA117, 12592–12594 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Guney, E. in Biocomputing 2017: Proceedings of the Pacific Symposium (eds Altmann, R. B. et al.) 132–143 (World Scientific, 2016).
- Xi, W. & Beer, M. A. Local epigenomic state cannot discriminate interacting and non-interacting enhancer-promoter pairs with high accuracy. PLoS Comput. Biol.14, e1006625 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Cao, F. & Fullwood, M. J. Inflated performance measures in enhancer-promoter interaction-prediction methods. Nat. Genet.51, 1196–1198 (2019). ArticleCASPubMedGoogle Scholar
- Whalen, S. & Pollard, K. S. Reply to ‘Inflated performance measures in enhancer-promoter interaction-prediction methods’. Nat. Genet.51, 1198–1200 (2019). ArticleCASPubMedGoogle Scholar
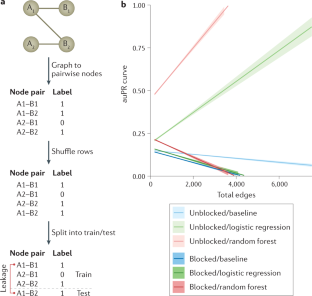
- Eid, F.-E. et al. Systematic auditing is essential to debiasing machine learning in biology. Commun. Biol.4, 183 (2020). This article proposes a set of data modifications that can be used to identify overestimated performance in supervised ML with paired-input data, such as protein–protein interactions, where examples occur in many pairs. ArticleGoogle Scholar
- Roberts, D. R. et al. Cross-validation strategies for data with temporal, spatial, hierarchical, or phylogenetic structure. Ecography40, 913–929 (2017). This study demonstrates blocking as an effective strategy for estimating the performance of ML models on data with complex dependency structures. ArticleGoogle Scholar
- Korte, A. et al. A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat. Genet.44, 1066–1071 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Stucki, S. et al. High performance computation of landscape genomic models including local indicators of spatial association. Mol. Ecol. Resour.17, 1072–1089 (2017). ArticleCASPubMedGoogle Scholar
- Runcie, D. E. & Crawford, L. Fast and flexible linear mixed models for genome-wide genetics. PLoS Genet.15, e1007978 (2019). ArticleCASPubMedPubMed CentralGoogle Scholar
- Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet.51, 1749–1755 (2019). ArticleCASPubMedGoogle Scholar
- Whalen, S., Truty, R. M. & Pollard, K. S. Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet.48, 488–496 (2016). ArticleCASPubMedPubMed CentralGoogle Scholar
- Brzyski, D. et al. Controlling the rate of GWAS false discoveries. Genetics205, 61–75 (2017). ArticlePubMedGoogle Scholar
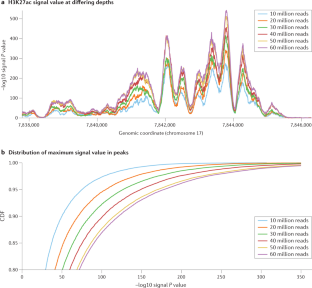
- Schreiber, J., Singh, R., Bilmes, J. & Noble, W. S. A pitfall for machine learning methods aiming to predict across cell types. Genome Biol.21, 282 (2020). ArticlePubMedPubMed CentralGoogle Scholar
- Lee, D., Redfern, O. & Orengo, C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol.8, 995–1005 (2007). ArticleCASPubMedGoogle Scholar
- Ribeiro, M. T., Singh, S. & Guestrin, C. in Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 1135–1144 (Association for Computing Machinery, 2016).
- Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc.7, 500–507 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Listgarten, J., Kadie, C., Schadt, E. E. & Heckerman, D. Correction for hidden confounders in the genetic analysis of gene expression. Proc. Natl Acad. Sci. USA107, 16465–16470 (2010). ArticleCASPubMedPubMed CentralGoogle Scholar
- Parsana, P. et al. Addressing confounding artifacts in reconstruction of gene co-expression networks. Genome Biol.20, 94 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Dinga, R., Schmaal, L., Brenda, W. J., Veltman, D. J. & Marquand, A. F. Controlling for effects of confounding variables on machine learning predictions. Preprint at bioRxivhttps://doi.org/10.1101/2020.08.17.255034 (2020).
- Dincer, A. B., Janizek, J. D. & Lee, S.-I. Adversarial deconfounding autoencoder for learning robust gene expression embeddings. Bioinformatics36, i573–i582 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Skafidas, E. et al. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol. Psychiatry19, 504–510 (2014). ArticleCASPubMedGoogle Scholar
- Robinson, E. B. et al. Response to ‘Predicting the diagnosis of autism spectrum disorder using gene pathway analysis’. Mol. Psychiatry19, 859–861 (2014). ArticleCASPubMedGoogle Scholar
- Keys, K. L. et al. On the cross-population generalizability of gene expression prediction models. PLoS Genet.16, e1008927 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Belgard, T. G., Jankovic, I., Lowe, J. K. & Geschwind, D. H. Population structure confounds autism genetic classifier. Mol. Psychiatry19, 405–407 (2014). ArticleCASPubMedGoogle Scholar
- Chen, X. et al. Drug-target interaction prediction: databases, web servers and computational models. Brief. Bioinform.17, 696–712 (2016). ArticleCASPubMedGoogle Scholar
- Brookhart, M. A., Stürmer, T., Glynn, R. J., Rassen, J. & Schneeweiss, S. Confounding control in healthcare database research: challenges and potential approaches. Med. Care48, S114–S120 (2010). ArticlePubMedPubMed CentralGoogle Scholar
- Zhang, J. M., Kamath, G. M. & Tse, D. N. Valid post-clustering differential analysis for single-cell RNA-seq. Cell Syst.9, 383–392.e6 (2019). ArticleCASPubMedPubMed CentralGoogle Scholar
- Gao, L. L., Bien, J. & Witten, D. Selective Inference for hierarchical clustering. Preprint at arXivhttps://arxiv.org/abs/2012.02936 (2020).
- Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res.12, 2825–2830 (2011). Google Scholar
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. Artic.28, 1–26 (2008). Google Scholar
- Vidaki, A. et al. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci. Int. Genet.28, 225–236 (2017). ArticleCASPubMedPubMed CentralGoogle Scholar
- Kimura, R. et al. An epigenetic biomarker for adult high-functioning autism spectrum disorder. Sci. Rep.9, 13662 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Levy, J. J. et al. MethylNet: an automated and modular deep learning approach for DNA methylation analysis. BMC Bioinforma.21, 108 (2020). ArticleCASGoogle Scholar
- Rauschert, S., Raubenheimer, K., Melton, P. E. & Huang, R. C. Machine learning and clinical epigenetics: a review of challenges for diagnosis and classification. Clin. Epigenetics12, 51 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature555, 469–474 (2018). ArticleCASPubMedPubMed CentralGoogle Scholar
- Bahado-Singh, R. O. et al. Deep learning/artificial intelligence and blood-based dna epigenomic prediction of cerebral palsy. Int. J. Mol. Sci.20, 2075 (2019). ArticleCASPubMed CentralGoogle Scholar
- Mohandas, N. et al. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin. Epigenetics10, 25 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Crowgey, E. L., Marsh, A. G., Robinson, K. G., Yeager, S. K. & Akins, R. E. Epigenetic machine learning: utilizing DNA methylation patterns to predict spastic cerebral palsy. BMC Bioinforma.19, 225 (2018). ArticleGoogle Scholar
- Aref-Eshghi, E. et al. Genomic DNA methylation-derived algorithm enables accurate detection of malignant prostate tissues. Front. Oncol.8, 100 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Luo, R. et al. Identifying CpG methylation signature as a promising biomarker for recurrence and immunotherapy in non-small-cell lung carcinoma. Aging12, 14649–14676 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Wilhelm-Benartzi, C. S. et al. Review of processing and analysis methods for DNA methylation array data. Br. J. Cancer109, 1394–1402 (2013). ArticleCASPubMedPubMed CentralGoogle Scholar
- Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin8, 6 (2015). ArticlePubMedPubMed CentralGoogle Scholar
- Rocke, D. M., Ideker, T., Troyanskaya, O., Quackenbush, J. & Dopazo, J. Papers on normalization, variable selection, classification or clustering of microarray data. Bioinformatics25, 701–702 (2009). ArticleCASGoogle Scholar
- Pulini, A. A., Kerr, W. T., Loo, S. K. & Lenartowicz, A. Classification accuracy of neuroimaging biomarkers in attention-deficit/hyperactivity disorder: effects of sample size and circular analysis. Biol. Psychiatry Cogn. Neurosci. Neuroimaging4, 108–120 (2019). PubMedGoogle Scholar
- Poldrack, R. A., Huckins, G. & Varoquaux, G. Establishment of best practices for evidence for prediction: a review. JAMA Psychiatry77, 534–540 (2020). ArticlePubMedPubMed CentralGoogle Scholar
- Ambroise, C. & McLachlan, G. J. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc. Natl Acad. Sci. USA99, 6562–6566 (2002). The authors present prediction of cancer outcome from expression of a small number of genes as an example of how supervised feature selection performed before cross-validation leads to performance overestimation. ArticleCASPubMedPubMed CentralGoogle Scholar
- van Eyk, C. L. et al. Analysis of 182 cerebral palsy transcriptomes points to dysregulation of trophic signalling pathways and overlap with autism. Transl. Psychiatry8, 88 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Alakwaa, F. M., Chaudhary, K. & Garmire, L. X. Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J. Proteome Res.17, 337–347 (2018). ArticleCASPubMedGoogle Scholar
- Yuan, Y., Guo, L., Shen, L. & Liu, J. S. Predicting gene expression from sequence: a reexamination. PLoS Comput. Biol.3, e243 (2007). ArticlePubMedPubMed CentralGoogle Scholar
- Urban, G., Torrisi, M., Magnan, C. N., Pollastri, G. & Baldi, P. Protein profiles: biases and protocols. Comput. Struct. Biotechnol. J.18, 2281–2289 (2020). This study demonstrates how protein profiles cause leakage of information between the training and test sets, and hence performance overestimation, in the context of protein structure prediction. ArticleCASPubMedPubMed CentralGoogle Scholar
- Khalilia, M., Chakraborty, S. & Popescu, M. Predicting disease risks from highly imbalanced data using random forest. BMC Med. Inform. Decis. Mak.11, 51 (2011). ArticlePubMedPubMed CentralGoogle Scholar
- Schubach, M., Re, M., Robinson, P. N. & Valentini, G. Imbalance-aware machine learning for predicting rare and common disease-associated non-coding variants. Sci. Rep.7, 2959 (2017). ArticlePubMedPubMed CentralGoogle Scholar
- Japkowicz, N. & Stephen, S. The class imbalance problem: a systematic study1. Intell. Data Anal.6, 429–449 (2002). ArticleGoogle Scholar
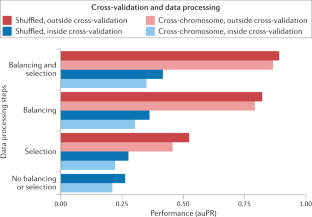
- Barandela, R., Sánchez, J. S., Garca, V. & Rangel, E. Strategies for learning in class imbalance problems. Pattern Recognit.36, 849–851 (2003). This work explores the negative consequences of imbalanced data as well as several common strategies for mitigating this pitfall. ArticleGoogle Scholar
- Batista, G. E. A. P. A., Prati, R. C. & Monard, M. C. A study of the behavior of several methods for balancing machine learning training data. SIGKDD Explor. Newsl.6, 20–29 (2004). ArticleGoogle Scholar
- Buda, M., Maki, A. & Mazurowski, M. A. A systematic study of the class imbalance problem in convolutional neural networks. Neural Netw.106, 249–259 (2018). This article explores performance measures and mitigation strategies for class imbalance specifically in the context of prediction with convolutional neural networks. ArticlePubMedGoogle Scholar
- Cui, Y., Jia, M., Lin, T.-Y., Song, Y. & Belongie, S. Class-balanced loss based on effective number of samples. In 2019 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR) (IEEE, 2019)
- Nguyen, H. M., Cooper, E. W. & Kamei, K. Borderline over-sampling for imbalanced data classification. Int. J. Knowl. Eng. Soft Data Paradig.3, 4 (2011). ArticleGoogle Scholar
- Chawla, N. V., Bowyer, K. W., Hall, L. O. & Kegelmeyer, W. P. SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res.16, 321–357 (2002). ArticleGoogle Scholar
- Haibo H., Yang B., Garcia, E. A. & Shutao L. in 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence) 1322–1328 (IEEE,2008).
- Lemaître, G., Nogueira, F. & Aridas, C. K. Imbalanced-learn: a python toolbox to tackle the curse of imbalanced datasets in machine learning. J. Mach. Learn. Res.18, 559–563 (2017). Google Scholar
- Davis, J. & Goadrich, M. in Proc. 23rd International Conference on Machine Learning 233–240 (Association for Computing Machinery, 2006).
- Peña-Castillo, L. et al. A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol.9, S2 (2008). ArticlePubMedPubMed CentralGoogle Scholar
- Kaler, A. S. & Purcell, L. C. Estimation of a significance threshold for genome-wide association studies. BMC Genomics20, 618 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics27, 1017–1018 (2011). ArticleCASPubMedPubMed CentralGoogle Scholar
- VanderWeele, T. J. & Shpitser, I. On the definition of a confounder. Ann. Stat.41, 196–220 (2013). ArticlePubMedPubMed CentralGoogle Scholar
- Efron, B. Prediction, estimation, and attribution. J. Am. Stat. Assoc.115, 636–655 (2020). ArticleCASGoogle Scholar
- Yu, B. & Kumbier, K. Veridical data science. Proc. Natl Acad. Sci. USA117, 3920–3929 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
Acknowledgements
The authors thank P. Baldi, M. Beer, A. Ben-Hur, J. Ernst, E. Eskin, G. Haliburton, H. Huang, S.-I. Lee, M. Libbrecht, J. Majewski, Q. Morris, S. Mostafavi, J.-P. Vert, W. Wang, B. Yu and M. Zitnik for recommending examples and for helpful suggestions on how to review this topic.
